Federal officials from the U. S. Food and Drug Administration (FDA) announced new restrictions Monday on the sale and distribution of Essure, the controversial permanent birth control device, but stopped short of removing it from the market.
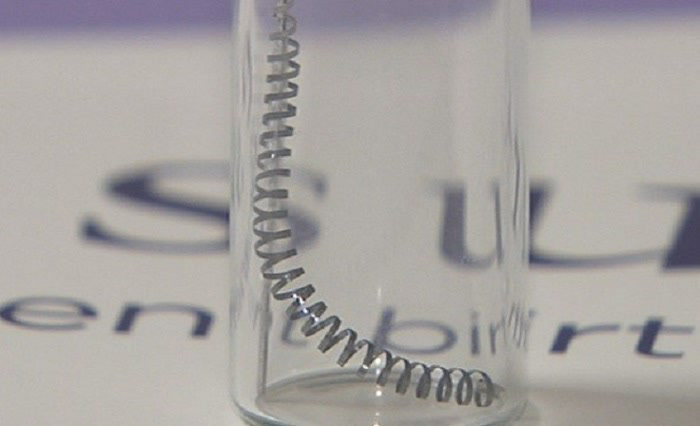
Essure is an implantable, permanent birth control device first approved by the FDA for use in 2002. The medical device is marketed as a safe, effective, low-cost, non-surgical alternative to surgical tubal ligation. The device consists of two small nickel-titanium coils that are inserted through the vagina and cervix and placed in the fallopian tubes. If the device works correctly, scar tissue develops around the fallopian tubes within about 3 months time, thereby preventing pregnancy by preventing fertilization.
Since arriving on the market in 2002, thousands of women have been injured by Essure and filed grievances with the FDA called Adverse Event Reports. Complaints include unrelenting severe bleeding, pain, perforated organs, unwarranted pregnancies, tubal pregnancies, depression, weight gain, and allergic reactions to the device, among other complaints.
After the FDA convened a panel of experts in September 2015 and opened public hearings for commentary, women injured by Essure hoped the device would be removed from the market but the FDA instead issued a tepid response in February 2016 requiring Bayer AG, maker of the device, to conduct post-market studies of the device, to add a black-box warning to the device, the strongest FDA warning required, and to create a patient checklist about the device’s risks.
In September 2017, Bayer stopped selling Essure worldwide outside the United States, and more than 12,000 new Adverse Event Reports were filed with the FDA in 2017, mostly by attorneys representing women injured by Essure describing their attempted removal of the device, resulting in hysterectomies and continuing problems.
When announcing new restrictions on the sale and distribution of the Essure implant on Monday, the FDA admitted it is imposing a “unique type of restriction” on Essure because of the continued problem with women being fully informed about the risks associated with the device.
FDA Commissioner Scott Gottlieb explained, “Despite previous efforts to alert women to the potential complications of Essure, we know that some patients still aren’t receiving this important information. That is simply unacceptable. Every single woman receiving this device should fully understand the associated risks.”
Healthcare facilities and healthcare providers approved to sell and distribute the device must present a detailed patient checklist brochure called the “Patient-Doctor Discussion Checklist – Acceptance of Risk and Informed Decision Acknowledgement” to each patient. Each patient and the doctor implanting the device must together review the 22-page brochure outlining all potential risks associated with the Essure implant, and both patient and doctor must sign off on the brochure.
The checklist is mandatory, and Bayer is solely responsible for implementing the new restrictions, and ensuring that providers comply. The FDA is threatening both civil and criminal penalties against Bayer if it fails to do both.
Although the thousands of women injured by Essure want the device off the market completely, they are satisfied for the moment that the FDA is finally taking their concerns seriously. “This is the first step the FDA has taken that actually benefits women over industry, and we are overjoyed to see that the Commissioner listened to our concerns.”
Bayer still contends the device is safe and effective despite the thousands of complaints, more than 16,000 lawsuits filed against by women injured by Essure, and the new FDA restrictions.